 SAEM
34ème édition de l'événement Société Algérienne d'Endocrinologie et Métabolisme
SAEM
34ème édition de l'événement Société Algérienne d'Endocrinologie et Métabolisme
 SPO
17ème édition de l'événement Société de Pédiatrie de l’Ouest
SPO
17ème édition de l'événement Société de Pédiatrie de l’Ouest
-
{{ result.title }}
{{ result.resume }}

Sous l'égide de
Avec le soutien de


Le syndrome de Leigh lié au gène NDUFA9 Etude clinique et génétique d’une famille
Moualek D (1), Saadi A (1), Bouderba R (1), Beghdadi Kh (2), Maroofian R (3), Houlden H (3), Nouioua S (1), Assami S (1), Tazir M (1), Ali Pacha L (1)
(1) Service de Neurologie CHU Mustapha Bacha- Alger
(2) Service de Radiologie CHU Mustapha Bacha- Alger
(3) Department of Neuromuscular Disorders, Department of Clinical and Experimental Epilepsy UCL Institute of Neurology, Queen Square- London
Introduction
Les troubles de la production d'énergie mitochondriale est le groupe le plus fréquent des troubles métaboliques héréditaires, avec une incidence estimée a au moins 1 sur 5000 naissances (1). La présentation clinique la plus courante étant le syndrome de Leigh (SL) avec une prévalence estimée à 1 pour 40 000 naissances (2).
Le SL également appelé encéphalopathie nécrosante subaiguë décrit pour la première fois en 1951 (3) se caractérise par des lésions cérébrales nécrotiques, bilatérales et symétriques en particulier des noyaux gris centraux et le tronc cérébral et une hétérogénéité clinique et génétique (4). Il débute dans la petite l’enfance mais peut se manifester aussi a l’ âge l’adulte.
Cliniquement le SL se manifeste plus fréquemment par une atteinte du système nerveux central (retard psychomoteur et ou régression psychomotrice, dystonie, convulsions, tremblements, nystagmus, ophtalmo parésie, atrophie optique, ataxie, et atteinte respiratoire) ou atteinte périphérique (neuropathie, myopathie) ou signes extra neurologiques (diabète, cardiomyopathie, anémie,…) (syndrome Leigh like) et une Hyperlactatemie dans le serum et le LCR (4).
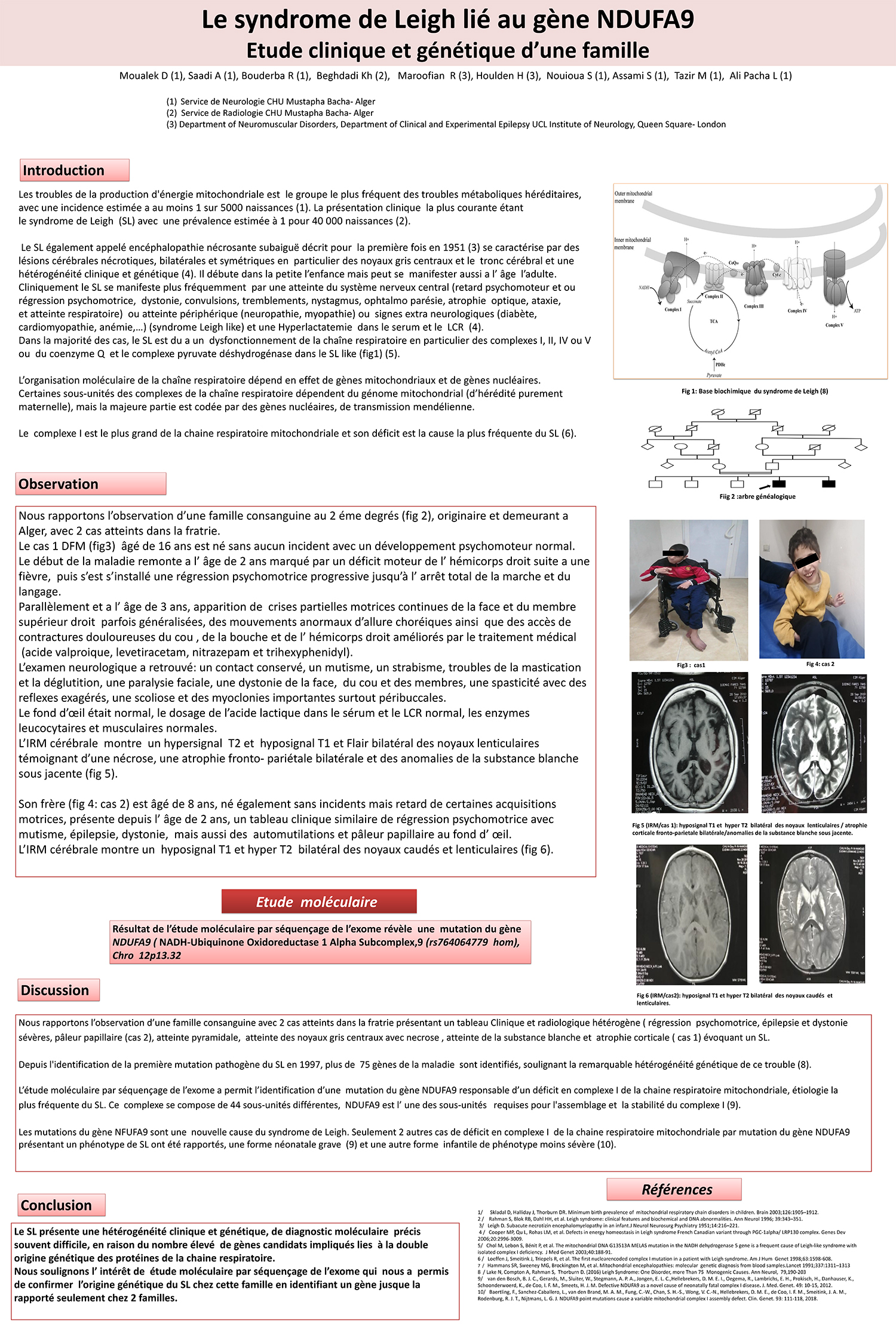
Dans la majorité des cas, le SL est du a un dysfonctionnement de la chaîne respiratoire en particulier des complexes I, II, IV ou V ou du coenzyme Q et le complexe pyruvate déshydrogénase dans le SL like (fig1) (5).
L’organisation moléculaire de la chaîne respiratoire dépend en effet de gènes mitochondriaux et de gènes nucléaires.
Certaines sous-unités des complexes de la chaîne respiratoire dépendent du génome mitochondrial (d’hérédité purement maternelle), mais la majeure partie est codée par des gènes nucléaires, de transmission mendélienne.
Le complexe I est le plus grand de la chaine respiratoire mitochondriale et son déficit est la cause la plus fréquente du SL (6).
Observation
Nous rapportons l’observation d’une famille consanguine au 2 éme degrés (fig 2), originaire et demeurant a Alger, avec 2 cas atteints dans la fratrie.
Le cas 1 DFM (fig3) âgé de 16 ans est né sans aucun incident avec un développement psychomoteur normal.
Le début de la maladie remonte a l’ âge de 2 ans marqué par un déficit moteur de l’ hémicorps droit suite a une fièvre, puis s’est s’installé une régression psychomotrice progressive jusqu’à l’ arrêt total de la marche et du langage.
Parallèlement et a l’ âge de 3 ans, apparition de crises partielles motrices continues de la face et du membre supérieur droit parfois généralisées, des mouvements anormaux d’allure choréiques ainsi que des accès de contractures douloureuses du cou , de la bouche et de l’ hémicorps droit améliorés par le traitement médical
(acide valproique, levetiracetam, nitrazepam et trihexyphenidyl).
L’examen neurologique a retrouvé: un contact conservé, un mutisme, un strabisme, troubles de la mastication et la déglutition, une paralysie faciale, une dystonie de la face, du cou et des membres, une spasticité avec des reflexes exagérés, une scoliose et des myoclonies importantes surtout péribuccales.
Le fond d’oeil était normal, le dosage de l’acide lactique dans le sérum et le LCR normal, les enzymes leucocytaires et musculaires normales.
L’IRM cérébrale montre un hypersignal T2 et hyposignal T1 et Flair bilatéral des noyaux lenticulaires témoignant d’une nécrose, une atrophie fronto- pariétale bilatérale et des anomalies de la substance blanche sous jacente (fig 5).
Son frère (fig 4: cas 2) est âgé de 8 ans, né également sans incidents mais retard de certaines acquisitions motrices, présente depuis l’ âge de 2 ans, un tableau clinique similaire de régression psychomotrice avec mutisme, épilepsie, dystonie, mais aussi des automutilations et pâleur papillaire au fond d’ oeil.
L’IRM cérébrale montre un hyposignal T1 et hyper T2 bilatéral des noyaux caudés et lenticulaires (fig 6).
Etude moléculaire
Résultat de l’étude moléculaire par séquençage de l’exome révèle une mutation du gène
NDUFA9 ( NADH-Ubiquinone Oxidoreductase 1 Alpha Subcomplex,9 (rs764064779 hom),
Chro 12p13.32
Discussion
Nous rapportons l’observation d’une famille consanguine avec 2 cas atteints dans la fratrie présentant un tableau Clinique et radiologique hétérogène ( régression psychomotrice, épilepsie et dystonie sévères, pâleur papillaire (cas 2), atteinte pyramidale, atteinte des noyaux gris centraux avec necrose , atteinte de la substance blanche et atrophie corticale ( cas 1) évoquant un SL.
Depuis l'identification de la première mutation pathogène du SL en 1997, plus de 75 gènes de la maladie sont identifiés, soulignant la remarquable hétérogénéité génétique de ce trouble (8).
L’étude moléculaire par séquençage de l’exome a permit l’identification d’une mutation du gène NDUFA9 responsable d’un déficit en complexe I de la chaine respiratoire mitochondriale, étiologie la plus fréquente du SL. Ce complexe se compose de 44 sous-unités différentes, NDUFA9 est l’ une des sous-unités requises pour l'assemblage et la stabilité du complexe I (9).
Les mutations du gène NFUFA9 sont une nouvelle cause du syndrome de Leigh. Seulement 2 autres cas de déficit en complexe I de la chaine respiratoire mitochondriale par mutation du gène NDUFA9 présentant un phénotype de SL ont été rapportés, une forme néonatale grave (9) et une autre forme infantile de phénotype moins sévère (10).
Conclusion
Le SL présente une hétérogénéité clinique et génétique, de diagnostic moléculaire précis souvent difficile, en raison du nombre élevé de gènes candidats impliqués lies à la double origine génétique des protéines de la chaine respiratoire.
Nous soulignons l’ intérêt de étude moléculaire par séquençage de l’exome qui nous a permis de confirmer l’origine génétique du SL chez cette famille en identifiant un gène jusque la rapporté seulement chez 2 familles.
Références
1/ Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 2003;126:1905–1912.
2 / Rahman S, Blok RB, Dahl HH, et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol 1996; 39:343–351.
3/ Leigh D. Subacute necrotizin encephalomyelopathy in an infant.J Neurol Neurosurg Psychiatry 1951;14:216–221.
4 / Cooper MP, Qu L, Rohas LM, et al. Defects in energy homeostasis in Leigh syndrome French Canadian variant through PGC-1alpha/ LRP130 complex. Genes Dev
2006;20:2996-3009.
5/ Chol M, Lebon S, Bénit P, et al. The mitochondrial DNA G13513A MELAS mutation in the NADH dehydrogenase 5 gene is a frequent cause of Leigh-like syndrome with
isolated complex I deficiency. J Med Genet 2003;40:188-91.
6 / Loeffen J, Smeitink J, Triepels R, et al. The first nuclearencoded complex I mutation in a patient with Leigh syndrome. Am J Hum Genet 1998;63:1598-608.
7 / Hammans SR, Sweeney MG, Brockington M, et al. Mitochondrial encephalopathies: molecular genetic diagnosis from blood samples.Lancet 1991;337:1311–1313
8 / Lake N, Compton A, Rahman S, Thorburn D. (2016) Leigh Syndrome: One Disorder, more Than 75 Monogenic Causes. Ann Neurol, 79,190-203
9/ van den Bosch, B. J. C., Gerards, M., Sluiter, W., Stegmann, A. P. A., Jongen, E. L. C.,Hellebrekers, D. M. E. I., Oegema, R., Lambrichs, E. H., Prokisch, H., Danhauser, K.,
Schoonderwoerd, K., de Coo, I. F. M., Smeets, H. J. M. Defective NDUFA9 as a novel cause of neonatally fatal complex I disease. J. Med. Genet. 49: 10-15, 2012.
10/ Baertling, F., Sanchez-Caballero, L., van den Brand, M. A. M., Fung, C.-W., Chan, S. H.-S., Wong, V. C.-N., Hellebrekers, D. M. E., de Coo, I. F. M., Smeitink, J. A. M.,
Rodenburg, R. J. T., Nijtmans, L. G. J. NDUFA9 point mutations cause a variable mitochondrial complex I assembly defect. Clin. Genet. 93: 111-118, 2018.
Dr. Dalila MOUALEK
Service de Neurologie CHU Mustapha Bacha- Alger
Association des Neurologues Privés d’Alger "ANPA"
webinaire - algérié , 2020-11-27 jusqu'a 2020-11-27
Santedz 2026 ©